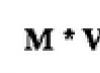Термодинамическим условием равновесия процесса, протекающего в изобарно-изотермических условиях, является равенство нулю изменения энергии Гиббса (D r G (Т )=0). При протекании реакции n а A + n b B = n с C+ n d Dизменение стандартной энергии Гиббса равно:
D r G 0 T =(n c ×D f G 0 C + n d ×D f G 0 D )–(n a ×D f G 0 A + n b ×D f G 0 B ).
Данное выражение соответствует идеальному процессу, в котором концентрации реагирующих веществ равны единице и неизменны в ходе реакции. В ходе реальных процессов концентрации реагентов меняются: концентрация исходных веществ уменьшается, а продуктов реакции увеличивается. С учетом концентрационной зависимости энергии Гиббса (см. химический потенциал) ее изменение в ходе реакции равно:
D r G T =–
– =
= (n c ×D f G 0 C + n d ×D f G 0 D ) – (n a ×D f G 0 A + n b ×D f G 0 B ) +
+ R×T ×(n c ×lnC C + n d ×lnC D – n a ×lnC A – n b ×lnC B )
D r G T = D r G 0 T + R×T × ,
где ![]() – безразмерная концентрация i
-го вещества;
– безразмерная концентрация i
-го вещества;
X i – мольная доля i -го вещества;
p i – парциальное давление i -го вещества; р 0 = 1,013×10 5 Па – стандартное давление;
с i – молярная концентрация i -го вещества; с 0 =1 моль/л – стандартная концентрация.
В состоянии равновесия
D r G
0 T + R×T
×  = 0,
= 0,
 .
.
Величина К 0 называется стандартной (термодинамичской) константой равновесия реакции. То есть при определенной температуре Т в результате протекания прямой и обратной реакции в системе устанавливается равновесие при определенных концентрациях реагирующих веществ – равновесных концентрациях (С i ) р . Величины равновесных концентраций определяются значением константы равновесия, которая является функцией температуры, и зависит от энтальпии (D r Н 0) и энтропии (D r S 0) реакции:
D r G 0 T + R ×T ×lnK 0 = 0,
![]() , ,
, ,
поскольку D r G 0 T =D r Н 0 T – Т ×D r S 0 T ,
![]() .
.
Если известны величины энтальпии (D r Н 0 T ) и энтропии (D r S 0 T ) или D r G 0 T реакции, то можно вычислить значение стандартной константы равновесия.
Константа равновесия реакции характеризует идеальные газовые смеси и растворы. Межмолекулярные взаимодействия в реальных газах и растворах приводят к отклонению расчетных величин констант равновесия от реальных. Для учета этого вместо парциальных давлений компонентов газовых смесей используется их фугитивность, а вместо концентрации веществ в растворах их активность (см. химический потенциал).
Сдвиг равновесия .
При равновесии в замкнутой системе устанавливаются равновесные концентрации реагирующих веществ. Если в системе изменяется один из параметров термодинамического равновесия (температура, давление, количество взаимодействующих веществ), то система переходит в другое состояние равновесия. Если в результате перехода равновесные концентрации продуктов реакции увеличиваются, то говорят о сдвиге равновесия в прямом направлении (вправо), если увеличиваются равновесные концентрации исходных веществ, то это сдвиг равновесия в обратном направлении (влево).
Определить «направление сдвига равновесия» можно при помощи уравнений изобары и изотермы реакции.
Изобара реакции
Производная lnK 0 по температуре при постоянном давлении равна:
![]() .
.
Данное уравнение называется изобарой реакции. На практике для приближенных расчетов можно считать, что D r Н 0 T » D r Н 0 298 , тогда
![]() .
.
Если известен знак теплового эффекта реакции, то можно определить «направление сдвига равновесия» при изменении температуры реакционной смеси.
Анализ уравнения изобары .
Пусть в системе протекает реакция
n а A + n b B↔ n с C + n d D.
![]() ,
,  .
.
Поскольку температура и универсальная газовая постоянная величины положительные, то знак производной функции lnK 0 (T )определяется знаком теплового эффекта реакции.
1. Экзотермическая реакция – D r Н 0 <0. Поскольку производная , то функция K (T ) убывающая, т. е. с увеличением температуры константа равновесия уменьшается. Следовательно, при возрастании температуры равновесие сдвигается в обратном направлении (уменьшение константы равновесия требует уменьшения числителя и соответственно увеличения знаменателя).
2. Эндотермическая реакция – D r Н 0 >0. Производная , следовательно, функция K (T ) возрастающая, т. е. с увеличением температуры константа равновесия увеличивается. При этом равновесие сдвигается в прямом направлении (увеличение константы равновесия требует увеличения числителя и уменьшения знаменателя).
Изотерма реакции
Пусть в системе протекает реакция n а A + n b B ↔ n с C + n d D. Если система не находится в состоянии равновесия (D r G T ¹0), то концентрации реагирующих веществ отличны от равновесных. В этом случае изменение энергии Гиббса реакции равно:
D r G T =D r G 0 T + R ×T × ¹ 0, D r G T =D r G 0 T + R ×T lnK Т × ¹0,
где ![]() – выражение, построенное по типу константы равновесия, содержащее концентрации реагирующих веществ в системе, не находящейся в состоянии равновесия. Эти концентрации в начальный момент времени являются произвольными и в ходе реакции изменяются до равновесных значений.
– выражение, построенное по типу константы равновесия, содержащее концентрации реагирующих веществ в системе, не находящейся в состоянии равновесия. Эти концентрации в начальный момент времени являются произвольными и в ходе реакции изменяются до равновесных значений.
Поскольку D r G 0 T + R ×T ×lnK 0 =0 ® D r G 0 T = – R ×T ×lnK 0 ,
где  – константа равновесия, то
– константа равновесия, то
D r G T = R ×T (lnK Т – lnK 0).
Это уравнение называется изотермой реакции . С его помощью можно определить направление протекания химической реакции при постоянной температуре в зависимости от соотношения концентраций реагентов.
Анализ уравнения изотермы .
1. Если соотношение концентраций исходных веществ (A, B) и продуктов (C, D) таково, что K Т = K 0 ,то D r G T = R ×T (lnK Т – lnK 0)=0. Система находится в состоянии равновесия.
2. Если соотношение исходных концентраций реагентов A, B, C и D таково, что K Т < K 0 , т. е.концентрация исходных веществ A и B больше равновесной, а концентрация продуктов C и D меньше, то D r G T = R ×T (lnK Т – lnK 0) <0. Реакция самопроизвольно протекает в прямом направлении. При этом концентрации исходных веществ уменьшаются, а продуктов увеличиваются. Соответственно увеличивается величина K Т . При достижении ею значения K 0 система приходит в состояние равновесия (D r G T =0).
3. Если соотношение исходных концентраций реагентов A, B, C и D таково, что K Т > K 0 , то изменение энергии Гиббса больше нуля. Реакция самопроизвольно протекает в обратном направлении до достижения системой состояния равновесия. При этом концентрации продуктов уменьшаются, а исходных веществ увеличиваются до равновесных значений.
Выводы о влиянии изменения температуры, давления и концентрации реагентов на смещение химического равновесия, сделанные при анализе уравнений изотермы и изобары реакции, находятся в полном соответствии с эмпирическим правилом Ле Шателье (Le Chatelier ). Если на систему, находящуюся в состоянии равновесия, оказать внешнее воздействие, то смещение равновесия происходит в сторону процесса, ослабляющего эффект внешнего воздействия. Это правило позволяет определять направление смещения равновесия.
Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
Размещено на http://www.allbest.ru/
Министерство образования Республики Беларусь
Учреждение образования
«Гомельский государственный университет
имени Франциска Скорины»
Биологический факультет
Кафедра Химии
У РС
Теория термодинамического равновесия
Выполнил
студент группы Би-31 А.Н. Коцур
Проверила С.М. Пантелеева
Гомель 2016
- 1. Различные виды равновесия
- 1.1 Неполное (Метастабильное) равновесие
- 1.2 Фазовое равновесие
- 1.3 Локальное термодинамическое равновесие
- 2. Критерии обратимости в качестве критериев равновесия
- 3. Некоторые условия устойчивости равновесия
- Список использованных источников
1 . Р азличные виды равновесия
1. 1 Неполное (Метастабильное) равновесие
В формулировке принципа необратимости говорится, что предельное (равновесное) состояние наступает с течением времени, рано или поздно, само собой, и что его признаком является прекращение всяких (не флуктуационных) изменений в системе. Легко, однако, привести примеры, когда это “с течением времени” растягивается до бесконечности, а система вообще не переходит “сама собой” в равновесное состояние, задерживаясь в каком-то другом состоянии, в котором так же не видно никаких изменений. Рассмотрим, например, газообразную смесь водорода и йода, адиабатически изолированную в закрытом сосуде. Количество атомов йода и атомов водорода можно взять произвольно. В предельном состоянии, в которое эта смесь должна перейти по принципу необратимости, все ее свойства должны однозначно определяться объемом сосуда, энергией смеси и количествами находящихся в ней атомов H и J. В частности, в предельном состоянии совершенно определенное количество атомов Н должно соединиться в молекулы Н 2 , совершенно определенное ко личество атомов J - в молекулы J 2 и должно получиться совершенно определенное количество молекул НJ. Следовательно, при приближении смеси к равновесию в ней должны идти реакции и т. д.
Однако если температура газа не очень высока, то такие превращения (например, диссоциация молекул Н 2) при столкновении частиц почти не происходят. Да и вообще, перегруппировка атомов в молекулах - процесс, часто идущий без катализаторов очень медленно и трудно. Поэтому в действительности, когда изменения в смеси прекратятся, в ней окажутся практически те же количества свободных атомов Н и J и те же количества молекул Н 2 , J 2 и НJ, которые имелись изначально, и в таком состоянии смесь может простоять очень долго. Она “задерживается” в состоянии, по существу, вовсе не равновесном, в чем можно убедиться, катализируя не идущие в ней реакции. Напри мер, если смесь осветить, то в ней начнется очень бурное, взрывное превращение молекул Н 2 и J 2 в НJ и смесь перейдет в новое “равновесие”, опять-таки неполное, поскольку реакция Н 2 2Н все равно еще не будет идти.
Если полное равновесие никогда не достигается, то сам принцип необратимости как будто теряет свой абсолютный характер; по- видимому, требуется новая его формулировка. Вопрос этот нельзя решить, не выяснив смысла понятия неполного равновесия. Если во обще различать равновесные (хотя бы и не вполне) и неравновесные состояния, то нужно понять, чем же они различаются. В чем прежде всего различие между полным и неполным равновесием? Неполное равновесие - это настоящее равновесие в системе, в которой некоторое свойство, способное меняться, когда нет за держивающих факторов, фиксировано. Величины, значения которых определяют какое-либо внутреннее свойство системы, часто называют внутренними параметрами. Можно сказать, что неполное равновесие - это настоящее равновесие в системе с фиксированными внутренними параметрами. Фиксирование внутренних параметров можно представить себе как результат действия некоторых дополнительных сил, под влиянием которых отдельные медленно идущие в системе процессы прекращаются вовсе. Конечно, такие силы вводятся только абстрактно. Система с фиксированными внутренними параметрами как будто становится другой системой - с другими внутренними движениями или с другим множеством микросостояний. Настоящее равновесие достигается тогда, когда нет никаких причин, мешающих внутренним движениям, и когда все идущие в системе процессы проходят до конца. Если же некоторые процессы протекают очень медленно и мы не дожидаемся их завершения или если какие-либо причины вообще прекращают отдельные внутренние процессы, то мы имеем дело как будто с новой системой, многообразие микросостояний которой меньше, чем у незаторможенной. В примере с газовой смесью роль внутренних параметров игра ют количества молекул Н 2 и J 2 . Состояния, в которых количества этих молекул отличаются от первоначальных, вовсе исключаются, так что молекулы Н 2 и J 2 рассматриваются как неделимые частицы. В примере с магнитом считается, что магнитные моменты отдельных доменов не могут меняться. Таким образом, мы высказываем следующее предположение: неполное равновесие является настоящим равновесием в системе с фиксированными внутренними параметрами. Чтобы его доказать, надо убедиться в применимости принципа необратимости к системам с фиксированными параметрами. Вряд ли есть основания сомневаться в этом. Однако нужно иметь в виду, что фиксирование внутренних параметров не должно быть таким, чтобы система фактически распалась на не связанные между собой части. Целесообразно различать случаи, когда скрытые движения совершенно не ограничены (в той мере, в какой это допускают фиксированные параметры), даже при неизменных механических параметрах отдельных частей системы, и случаи, когда отдельные части системы вообще изолированы друг от друга или могут передавать друг другу движение только при изменении механических параметров отдельных частей, т. е. через посредство механических систем. В первом случае мы будем называть систему термически однородной, а во втором - термически неоднородной . Термически однородная система с фиксированными параметрами полностью подчиняется принципу необратимости и переходит при неизменных внешних условиях в предельное состояние, которое будет для нее настоящим равновесием; для системы со свободными внутренними параметрами подобное состояние является неполным равновесием. Это неполное равновесие не зависит от начального состояния системы, если фиксированные параметры вначале имели нужные (фиксированные) значения. В неполном равновесии также не остается никакого следа от приведшего к нему процесса. Например, смесь определенных количеств молекул Н 2 и J 2 можно взять в данном объеме и с данной энергией в самых разнообразных начальных состояниях: молекулы смеси можно произвольно разместить в объеме, между ними можно самыми разнообразными способами распределить энергию. Окончательное (неполное) равновесие (равновесие при неизменных количествах молекул Н 2 и J 2) будет всегда одно и то же. Поскольку любое микросостояние рассматриваемой системы с заданными количествами Н2 и J 2 может перейти в любое другое такое микросостояние, система термически однородна. Для термически неоднородных систем принцип необратимости не имеет места, и понятно почему. Энергия каждой части такой системы может и не быть фиксирована. Предполагается, что энергия любой части меняется только при изменении ее механических параметров. Однако если силы, действующие со стороны нескольких частей системы вдоль этих параметров, в сумме равны нулю (уравновешиваются), то параметры остаются неизменными. Тогда энергия рассматриваемой части системы будет постоянной и в ней наступит равновесие, определяемое значениями ее механических параметров и ее энергией. Но эти энергия (при данной общей энергии системы) и значения механических параметров (при данных значениях внешних для всей системы механических параметров) могут быть разными; тогда вся система будет иметь несколько равновесий при одних и тех же внешних условиях и одной и той же энергии.
равновесие термодинамический изобарный
1. 2 Фазовое равновесие
Фазовое равновесие, одновременное существование термодинамически равновесных фаз в многофазной системе. Простейшие примеры - равновесие жидкости со своим насыщенным паром, равновесие воды и льда при температуре плавления, расслоение смеси воды с триэтиламином на два несмешивающихся слоя (две фазы), отличающихся концентрациями. В равновесии могут находиться (в отсутствии внешнего магнитного поля) две фазы ферромагнетика с одинаковой осью намагничивания, но различным направлением намагниченности; нормальная и сверхпроводящая фазы металла во внешнем магнитном поле и т.д. При переходе в условиях равновесия частицы из одной фазы в другую энергия системы не меняется. Другими словами, при равновесии химические потенциалы каждой компоненты в различных фазах одинаковы. Отсюда следует фаз правилоГиббса: в веществе, состоящем из k компонент, одновременно могут существовать не более чем k + 2 равновесные фазы. Например, в однокомпонентном веществе число одновременно существующих фаз не превосходит трёх (см. Тройная точка).Число термодинамических степеней свободы, т. е. переменных (физических параметров), которые можно изменять, не нарушая условий Фазовое равновесие, равно
где j - число фаз, находящихся в равновесии.
Например, в двухкомпонентной системе три фазы могут находиться в равновесии при разных температурах, но давление и концентрации компонент полностью определяются температурой. Изменение температуры фазового перехода (кипения, плавления и др.) при бесконечно малом изменении давления определяется Клапейрона - Клаузиуса уравнением. Графики, изображающие зависимость одних термодинамических переменных от других в условиях Фазовое равновесие, называются линиями (поверхностями) равновесия, а их совокупность - диаграммами состояния. Линия Фазовое равновесие может либо пересечься с другой линией равновесия (тройная точка), либо кончиться критической точкой.
В твёрдых телах из-за медленности процессов диффузии, приводящих к термодинамическому равновесию, возникают неравновесные фазы, которые могут существовать наряду с равновесными. В этом случае правило фаз может не выполняться. Правило фаз не выполняется также и в том случае, когда на кривой равновесия фазы не отличаются друг от друга (см. Фазовые переходы).
В массивных образцах в отсутствии дальнодействующих сил между частицами число границ между равновесными фазами минимально. Например, в случае двухфазного равновесия имеется лишь одна поверхность раздела фаз. Если хотя бы в одной из фаз существует дальнодействующее поле (электрическое или магнитное), выходящее из вещества, то энергетически более выгодны равновесные состояния с большим числом периодически расположенных фазовых границ (домены ферромагнитные и сегнетоэлектрические, промежуточное состояние сверхпроводников) и таким расположением фаз, чтобы дальнодействующее поле не выходило из тела. Форма границы раздела фаз определяется условием минимальности поверхностной энергии. Так, в двухкомпонентной смеси при условии равенства плотностей фаз граница раздела имеет сферическую форму. Огранка кристаллов определяется теми плоскостями, поверхностная энергия которых минимальна.
1.3 Локальное термодинамическое равновесие
Одно из основных понятий термодинамики неравновесных процессов и механики сплошных сред; равновесие в очень малых (элементарных) объёмах среды, содержащих всё же столь большое число частиц (молекул, атомов, ионов и др.), что состояние среды в этих физически бесконечно малых объёмах можно характеризовать темп-рой Т (х), хим. потенциалами(х)и др. термодинамические параметрами, но не постоянными, как при полном равновесии, а зависящими от пространств, координат х и времени. Ещё один параметр Л.Т.Р.- гидродинамическая скорость и (х) - характеризует скорость движения центра масс элемента среды. При Л.Т.Р. элементов среды состояние среды в целом неравновесно. Если малые элементы среды рассматривать приближённо как термодинамически равновесные подсистемы и учитывать обмен энергией, импульсом и веществом между ними на основе уравнений баланса, то задачи термодинамики неравновесных процессов решаются методами термодинамики и механики. В состоянии Л.Т.Р. плотность энтропии s(z)на единицу массы является функцией плотности внутренней энергии и концентраций компонентов Сk (x), такой же, как и в состоянии равновесия термодинамического. Термодинамического равенства остаются справедливыми для элемента среды при движении вдоль пути его центра масс:
где grad, (х)- давление, - удельный объём.
Статистическая физика позволяет уточнить понятие Л.Т.Р. и указать пределы его применимости. Понятию Л.Т.Р. соответствует локально равновесная функция распределения f плотности энергии, импульса и массы, которая отвечает максимуму информационной энтропии при заданных средних значениях этих величин как функций координат и времени:
где Z - статистическая сумма, (х) - динамическая переменные (функции координат и импульсов всех частиц системы), соответствующие плотности энергии (в системе координат, движущейся с гидродинамической скоростью) и плотности массы. При помощи такой функции распределения можно определить понятие энтропии неравновесного состояния как энтропии такого локально равновесного состояния, которое характеризуется теми же значениями плотностей энергии, импульса и массы, что и рассматриваемое неравновесное состояние. Однако локально равновесное распределение позволяет получать лишь уравнения т. н. идеальной гидродинамики, в которых не учитываются необратимые процессы. Для получения уравнений гидродинамики, учитывающих необратимые процессы теплопроводности, вязкости и диффузии (т. е. переноса явления), требуется обращаться к кинетическому уравнению для газов или к Лиувилля уравнению, справедливому для любой среды, и искать такие их решения, которые зависят от координат и времени лишь через средние значения параметров, определяющих неравновесное состояние. В результате получается неравновесная функция распределения, которая позволяет вывести все уравнения, описывающие процессы переноса энергии, импульса и вещества (уравнения диффузии, теплопроводности и Навье - Стокса уравнения) .
2. Критерии обратимости в качестве критериев равновесия
Пользуясь тем, что п изохорно-изотермическом обратимом процессе d ut U = Td ut S . Выведем критерии равновесия произвольной термодинамической системы, основываясь, на том, что равновесие - необходимое условие обратимости процесса и что, таким образом, каждое из состояний, через которые проходит система в обратимом процессе, оказывается состоянием равновесия. Отсюда следует: Критерии обратимости всегда являются вместе с тем критериями равновесия. Этим обстоятельством и пользуются в термодинамике: определяют состояния, в которых может происходить обратимый процесс, н каждое такое состояние считают состоянием равновесия. В настоящее время в термодинамике нет других средств нахождения состояний равновесия. Однако, пользуясь критериями обратимости вместо критериев равновесия, нужно помнить, что равновесие- необходимое, но недостаточное условие обратимости, т. с, что, кроме равновесных состояний, в которых может начаться обратимый процесс, существуют и такие равновесные состояния, в которых обратимый процесс невозможен. Из этого явствует, что, применяя критерии обратимости в качестве критериев равновесия, можно определить не все состояния равновесия, а только часть их. Этим объясняется тот хорошо известный факт, что все предсказанные термодинамикой состояния равновесия, действительно имеют место; но, кроме них, наблюдаются и такие состояния, которые термодинамикой не предсказываются. Между тем в некоторых таких смесях в довольно значительном интервале температур при постоянном объеме равновесный состав также остается постоянным, т. е. имеется непрерывный ряд равновесий и только одно из них указывается термодинамикой .
3. Некоторые условия устойчивости равновесия
Специальный термодинамический анализ позволяет показать, что из соображений термодинамической устойчивости системы для любого вещества должны выполняться следующие соотношения:
т.е., во-первых, изохорная теплоемкость С v всегда положительна и, во-вторых, в изотермическом процессе увеличение давления всегда приводит к уменьшению объема вещества. Условие (1) называют условием термической устойчивости, а условие (2) - условием механической устойчивости. Условия (1) и (2) можно объяснить так называемым принципом смещения равновесия (принцип Ле Шателье - Брауна), смысл которого заключается в том, что, если система, находившаяся в равновесии, выводится из него, соответствующие параметры системы изменяются таким образом, чтобы система вернулась в состояние равновесия. Эти условия термодинамической устойчивости системы ясны и без формальных выкладок. Представим себе, что теплоемкость сv некоторого вещества отрицательна. Это означало бы, поскольку cv = dq v /dT , что подвод теплоты к веществу при постоянном объеме этого вещества приводил бы не к повышению, а к понижению температуры. Таким образом, чем больше теплоты мы подводили бы к веществу в изохорном процессе, тем больше становилась бы разность между температурами этого вещества и источника теплоты (окружающая среда).
Для вывода условий устойчивости можно предположить, что при малом отклонении от положения равновесия система однородна по внутренним параметрам T иp, ноTT o ,PP o , пока не достигнуто равновесие. Можно обойтись и без этого предположения и рассмотреть не всю систему, а столь малую ее часть, что ее можно считать однородной поTиp. Результат будет получен один и тот же. Согласно (49) запишем
dU-T c dS+p c dV=-T c (d i S+d i S пов )
Если система выведена из условия устойчивого равновесия, то поскольку правая часть положительна, то
dU-T c dS+p c dV>0.
При малом, но не бесконечно малом отклонении от устойчивого равновесия должно быть
U-T c S+p c V>0 (51)
При этом U=T S-p V . Подставляя это выражение в (51) получим условия устойчивости равновесия в виде
TS-pV>0, (52)
где T=T-T c ,p=p-p c отклоненияT иp от равновесных значений поскольку в равновесииT=T c , p=p c .
Для изобарных (p=0) и изохорных (V=0) систем условия устойчивости равновесия (52) принимают видTS>0
Будем неограниченно приближать систему к равновесию, меняя S. Тогда
В изобарных и изохорных условиях
Следовательно, условие устойчивости изобарного равновесия имеет вид, (53)то есть,. (54)
Условие устойчивости изохорного равновесия, (55) то есть, . (56)
В изотермической (T=0)и изэнтропической (S=0)системах условие (52) принимает видpV<0. Будем неограниченно приближать систему к равновесию, меняя V. Тогда
в изотермических, а в изэнтропических условиях
Следовательно, условие устойчивости изотермического равновесия имеет вид. То есть(57) или T >0 (58)
Для изэнтропического равновесия - , то есть, (59) или S >0(60)
Неравенства,называют условиями термической устойчивости, а неравенства, T >0, S >0 называют условием механической устойчивости равновесия системы. Равновесие изобарно-изотермической системы устойчиво при одновременном выполнении как условия термической (54), так и механической устойчивости (58) T >0. Физический смысл условий устойчивости ясен из их вывода. Термодинамическое равновесие термически устойчиво, если термические флуктуации (отклонения от равновесного значения энтропииSприT=constили температурыTприS=consrt)выводят систему в такое неравновесное состояние, из которого она возвращается в исходное равновесное состояние. Термодинамическое состояние механически устойчиво, если“механические”флуктуации (отклонения от равновесного объемаVприp=const или давленияPприV=const) выводят систему в такое неравновесное состояние, из которого она возвращается в исходное равновесное.
Термодинамическое равновесие неустойчиво, если сколь угодно малые флуктуации выводят систему в такое неравновесное состояние, из которого она не возвращается в исходное равновесное, а движется к некоторому иному равновесному.
Следует отметить, что, если в данных условиях рассматриваемое равновесное состояние оказывается неустойчивым (не выполнены условия устойчивости), то при этих условиях существует непременно некоторое иное, устойчиво равновесное состояние. Система не может находиться в неустойчивом равновесии сколь - нибудь долго. Понятие неустойчиво равновесное состояния достаточно условно. Строго говоря, неустойчиво равновесные состояния не реализуются. Могут существовать лишь неравновесные состояния, в какой-то мере близкие или приближающиеся к неустойчиво равновесным.
Если выполнены все условия устойчивости (54),(56),(57),(58), то все четыре характеристик C P ,C V , S T положительны. При этом,как видно, из (43)C P >C V и, как следует из (37) T > S .
Как видно из (36), P может быть и положительным и отрицательным; знак P не определяется условиями устойчивости, Из опыта известно, что почти всегда P >0. При этом, как следует из (39) и (40) изохорный и адиабатический коэффициенты давленияпри выполнении условий устойчивости V >0, S >0. Если выполнены условия C P >0, T >0, то из (41) следует P > S и, вообще говоря, P и S могут иметь разный знак.
Список использованных источников
1Сорокин, В. С. Макроскопическая необратимость и энтропия. Введение в термодинамику. / В.С. Сорокин. - М.: ФИЗМАТЛИТ, 2004. - 176 с.
2Михеева, Е.В. Физическая и коллоидная химия: учебное пособие / Е.В.Михеева, Н.П.Пикула; Томский политехнический университет. - Томск: ТПУ, 2010. - 267 с.
3Де Гроот, С. Неравновесная термодинамика. / С. Де Гроот, П. Мазур. М.: Мир, 1964. - 456 с.
4Химия и химическая технология / Некоторые условия устойчивости равновесия [Электронный ресурс] // URL: http://www.chem21.info/page/104.html (дата обращения 18.04.2016).
Размещено на Allbest.ru
...Подобные документы
Термодинамико-топологический анализ структур диаграмм фазового равновесия. Закономерности векторного поля нод и скалярного поля равновесных температур. Уравнение их взаимосвязи. Нелокальные закономерности диаграмм фазового равновесия жидкость – пар.
дипломная работа , добавлен 04.01.2009
Рассчет сродства соединений железа к кислороду воздуха при определееной константе равновесия реакции. Определение колличества разложившегося вещества при нагревании. Вычисление константы равновесия реакции CO+0,5O2=CO2 по стандартной энергии Гиббса.
тест , добавлен 01.03.2008
Характеристика химического равновесия в растворах и гомогенных системах. Анализ зависимости константы равновесия от температуры и природы реагирующих веществ. Описания процесса синтеза аммиака. Фазовая диаграмма воды. Исследование принципа Ле Шателье.
презентация , добавлен 23.11.2014
Современное состояние исследований в области азеотропии. Термодинамико-топологический анализ структур диаграмм парожидкостного равновесия. Новый подход к определению классов диаграмм трехкомпонентных биазеотропных систем. Математическое моделирование.
дипломная работа , добавлен 12.11.2013
Характеристика химического равновесия. Зависимость скорости химической реакции от концентрации реагирующих веществ, температуры, величины поверхности реагирующих веществ. Влияние концентрации реагирующих веществ и температуры на состояние равновесия.
лабораторная работа , добавлен 08.10.2013
Получение и применение силицидов марганца. Химические и фазовые равновесия в системе Mn-Si. Обобщенная теория "регулярных" растворов. Термодинамические функции образования интерметаллидов. Интерполяционная формула Лагранжа. Формулы Миедемы и Истмена.
дипломная работа , добавлен 13.03.2011
Расчетные методы определения рН. Примеры уравнений реакций гидролиза солей. Понятие и формулы расчета константы и степени гидролиза. Cмещение равновесия (вправо, влево) гидролиза. Диссоциация малорастворимых веществ и константа равновесия этого процесса.
лекция , добавлен 22.04.2013
Определение константы равновесия реакции. Вычисление энергии активации реакции. Осмотическое давление раствора. Схема гальванического элемента. Вычисление молярной концентрации эквивалента вещества. Определение энергии активации химической реакции.
контрольная работа , добавлен 25.02.2014
Понятие и единицы измерения адсорбции. Зависимость величины адсорбции от концентрации, давления и температуры. Изотерма, изобара, изопикна, изостера адсорбции. Поверхностно-активные и поверхностно-инактивные вещества. Уравнения адсорбционного равновесия.
реферат , добавлен 22.01.2009
Понятие химического анализа. Теоретические основы количественного химического анализа. Требования к химическим реакциям. Понятие и суть эквивалента вещества. Понятие химического равновесия и законы действующих масс. Константы равновесия реакций и их суть.
Термодинамическое условие химического равновесия
Термодинамическим условием равновесия процесса, протекающего в изобарно-изотермических условиях, является равенство нулю изменения энергии Гиббса (D r G (Т )=0). При протекании реакции n а A + n b B = n с C + n d D
изменение стандартной энергии Гиббса равно˸
D r G 0 T =(n c ×D f G 0 C + n d ×D f G 0 D )–(n a ×D f G 0 A + n b ×D f G 0 B ).
Данное выражение соответствует идеальному процессу, в котором концентрации реагирующих веществ равны единице и неизменны в ходе реакции. В ходе реальных процессов концентрации реагентов меняются˸ концентрация исходных веществ уменьшается, а продуктов реакции увеличивается. С учетом концентрационной зависимости энергии Гиббса (см. п. 1. 3. 4) её изменение в ходе реакции равно˸
D r G T =–
– =
=(n c ×D f G 0 C + n d ×D f G 0 D )–(n a ×D f G 0 A + n b ×D f G 0 B ) +
+ R ×T ×(n c ×lnC C + n d ×lnC D –n a ×lnC A –n b ×lnC B )
D r G T =D r G 0 T + R ×T × ,
где – безразмерная концентрация i -го вещества; X i – мольная доля i -го вещества; p i – парциальное давление i -го вещества; р 0 = =1,013×10 5 Па – стандартное давление; с i – молярная концентрация i -го вещества; с 0 =1 моль/л – стандартная концентрация.
В состоянии равновесия
D r G 0 T + R×T × = 0,
Величина К 0 называется стандартной (термодинамичской) константой равновесия реакции. Таким образом при определенной температуре Т в результате протекания прямой и обратной реакции в системе устанавливается равновесие при определенных концентрациях реагирующих веществ – равновесных концентрациях (С i ) р . Величины равновесных концентраций определяются значением константы равновесия, которая является функцией температуры, и зависит от энтальпии (D r Н 0) и энтропии (D r S 0) реакции˸
D r G 0 T + R ×T ×lnK 0 = 0,
поскольку D r G 0 T =D r Н 0 T – Т ×D r S 0 T ,
Если известны величины энтальпии (D r Н 0 T ) и энтропии (D r S 0 T ) или D r G 0 T реакции, то можно вычислить значение стандартной константы равновесия.
Константа равновесия реакции характеризует идеальные газовые смеси и растворы. Межмолекулярные взаимодействия в реальных газах и растворах приводят к отклонению расчетных величин констант равновесия от реальных. Для учета этого вместо парциальных давлений компонентов газовых смесей используется их фугитивность, а вместо концентрации веществ в растворах их активность. Фугитивность i -го компонента связана с ᴇᴦο парциальным давлением соотношением f i =g i ×p i , где g i – коэффициент фугитивности.Активность и концентрация компонента связаны соотношением а i =g i ×С i , где g i – коэффициент активности.
Необходимо отметить, что в достаточно широкой области давлений и температур газовые смеси можно считать идеальными и проводить расчёты равновесного состава газовой смеси, считая коэффициент фугитивности g i @ 1.В случае жидких растворов, особенно растворов электролитов, коэффициенты активности их компонентов могут значительно отличаться от единицы (g i ¹ 1) и для расчета равновесного состава необходимо использовать активности.
Термодинамическое условие химического равновесия - понятие и виды. Классификация и особенности категории "Термодинамическое условие химического равновесия" 2015, 2017-2018.
1. Экстремальные свойства термодинамических потенциалов.
2. Условия равновесия и устойчивости пространственно однородной системы.
3. Общие условия равновесия фаз в термодинамических системах.
4. Фазовые переходы I-го рода.
5. Фазовые переходы II-го рода.
6. Обобщение полуфеноменологической теории.
Вопросы устойчивости термодинамических систем рассматривались в предыдущей теме применительно к задаче химического равновесия. Поставим задачу теоретического обоснования сформулированных ранее условий (3.53) на основе II начала термодинамики, используя свойства термодинамических потенциалов.
Рассмотрим макроскопическое бесконечно малое изменение состояния системы: 1 -2, при котором все ее параметры относятся на бесконечно малую величину:
Соответственно:
Тогда в случае квазистатического перехода из обобщенной формулировки I и II начала термодинамики (2.16) следует:
В случае, если 1-2 является неквазистатическим, то выполняются следующие неравенства:
В выражении (4.3) величины со штрихом соответствуют неквазистатическому процессу, а величины без штриха - квазистатическому. Первое неравенство системы (4.3) характеризует полученный на основе обобщения многочисленных опытных данных принцип максимального поглощения тепла, а второе - принцип максимальной работы.
Записывая работу для неквазистатического процесса в виде и вводя аналогичным образом параметры и, получим:
Выражение (4.4) абсолютно эквивалентно неравенству Клаузиуса.
Рассмотрим основные следствия (4.4) для различных способов описания термодинамических систем:
1. Адиабатически изолированная система: (). Соответственно. Тогда:
Это означает, что если зафиксировать переменные состояния системы, то вследствие (4.5) ее энтропия будет возникать до тех пор, пока в системе, согласно нулевого начала термодинамики, не наступит состояния равновесия. То есть равновесия состояния соответствует максимуму энтропии:
Вариации в (4.6) производятся по тем параметрам, которые при указанных фиксированных параметрах системы могут принимать неравновесные значения. Это могут быть концентрация п , давление р , температура ит.д.
2. Система в термостате (). Соответственно что позволяет переписать (4.4) в виде:
Учитывая вид выражения для свободной энергии: и равенство, получаем:
Таким образом течение неравновесных процессов для системы, помещенной в термостат, сопровождается уменьшением ее свободной энергии. А равновесное значенте соответствует ее минимуму:
3. Система под поршнем (), т.е. .В этом случае соотношение (4.4) принимает вид:
Таким образом равновесие в системе под поршнем наступает при достижении минимального значения потенциала Гиббса:
4. Система с воображаемыми стенками (). Тогда. Тогда
что позволяет записать
Соответственно в системе с воображаемыми стенками неравновесные процессы направлены в сторону уменьшения потенциала, а равновесие достигается при условии:
Условие определяет само состояние равновесия системы и широко используется при исследовании многокомпонентных или многофазных систем. Условия минимума или максимума определяют критерии устойчивости этих равновесных состояний по отношению к самопроизвольным или искусственно создаваемым возмущениям системы.
Кроме того, наличие экстремальных свойств у термодинамических потенциалов позволяет использовать для их исследования вариационных методов по аналогии с вариационными принципами механики. Однако, в этих целях требуется использование статистического подхода.
Рассмотрим условия равновесия и устойчивости термодинамических систем на примере газа, помещенного в цилиндр над поршнем. Кроме того, для упрощения анализа пренебрежем внешними полями, полагая. Тогда переменными состояния являются ().
Ранее отмечалось, что на термодинамическую систему можно оказывать воздействия либо совершая работу над ней, либо сообщая ей некоторое количество тепла. Поэтому следует проанализировать равновесие и устойчивость по отношению к каждому из отмеченных воздействий.
Механическое воздействие связано со смещением незакрепленного поршня. В этом случае работа на систему равно
В качестве внутреннего параметра, который может изменяться и по которому следует осуществлять варьирование, выберем объем.
Представляя потенциал Гиббса через свободную энергию
и производя варьирование, запишем:
Из последнего равенства следует:
Выражение (4.13) следует рассматривать как уравнение относительно равновесного значения объема при заданных параметрах системы ().
Условия устойчивости равновесного состояния имеет вид:
Учитывая (4.13), последнее условие можно переписать в виде:
Условие (4.14) накладывает определенные требования на уравнение состояния. Так, изотермы идеального газа
всюду удовлетворяют условию устойчивости. В то же время, уравнение Ван-дер-Ваальса
или уравнения Дитериги
имеют участки на которых условия устойчивости не выполняются, и которые не соответствуют реальным равновесным состояниям, т.е. экспериментально реализуется.
Если же в некоторой точке изотермы, то для проверки устойчивости используют специальные методы математического анализа, т.е. проверяют выполнение условий:
Аналогичным образом требования устойчивости, предъявляемые к уравнению состояния, могут быть сформулированы и для других параметров системы. Рассмотрим в качестве примера зависимость химического потенциала. Введем плотность числа частиц. Тогда химический потенциал можно представить в виде.
Вычислим дифференциал в зависимости от переменных состояния:
При записи последнего выражения учтено, что и использовано термодинамическое тождество (3.8). Тогда
То есть условие устойчивости для химического потенциала принимает вид
В критической точке при наличии прогиба имеем:
Перейдем к анализу устойчивости системы к тепловому воздействию, связанного с передачей некоторого количества тепла. Тогда в качестве вариационного параметра рассмотрим энтропию системы S . Для учета именно теплового воздействия зафиксируем механические параметры. Тогда в качестве переменных термодинамического состояния удобно выбрать набор, а в качестве термодинамического потенциала свободную энергию.
Выполняя варьирование, находим:
Из условия равновесия получаем
Уравнения (4.21) следует рассматривать как уравнение для равновесного значения энтропии. Из положительности второй вариации свободной энергии:
Поскольку температура всегда принимает положительные значения из (4.22) следует:
Выражение (4.23) является искомым условием устойчивости термодинамической системы по отношению к нагреванию. Некоторые авторы рассматривают положительность теплоемкости как одно из проявлений принципа Ле-Шателье - Брауна. При сообщении термодинамической системе количества тепла:
Ее температура возникает, что, в соответствии со вторым началом термодинамики в формулировке Клаузиуса (1850г.), приводит к уменьшению количества теплоты, поступающего в систему. Иначе говоря, в ответ на внешние воздействия - сообщение количества теплоты - термодинамические параметры системы (температура) меняются таким образом, что внешние воздействия ослабляются.
Рассмотрим вначале однокомпонентную систему, находящуюся в двухфазном состоянии. Здесь и далее под фазой будем понимать однородное вещество в химическом и физическом отношении.
Таким образом, каждую фазу будем рассматривать как однородную и термодинамически устойчивую подсистему, характеризуемую общим значением давления (в соответствии с требованием отсутствия тепловых потоков). Исследуем условие равновесия двуфазной системы по отношению к изменению числа частиц и, находящихся в каждой из фаз.
С учетом сделанных допущений наиболее удобным является использование описания системы под поршнем с фиксацией параметров (). Здесь - общее число частиц в обеих фазах. Также для простоты “выключим” внешние поля (а =0).
В соответствии с выбранным способом описания условием равновесия является условие (4.10) минимума потенциала Гиббса:
которое дополняется условием постоянства числа частиц N :
Выполняя варьирование в (4.24а) с учетом (4.24б) находим:
Таким образом, общим критерием равновесия двуфазной системы является равенство их химических потенциалов.
Еси известны выражения химических потенциалов и, то решением уравнения (4.25) будет некоторая кривая
называемая кривой фазового равновесия или дискретной фазового равновесия.
Зная выражения для химических потенциалов, из равенства (2.юю):
мы можем найти удельные объемы для каждой из фаз:
То есть, (4.26) можно переписать в виде уравнений состояния для каждой из фаз:
Обобщим полученные результаты на случай n фаз и k химически нереагирующих компонент. Для произвольной i -й компоненты уравнение (4.25) примет вид:
Легко видеть, что выражение (4.28) представляет систему (n- 1) независимых уравнений. Соответственно из условий равновесия для k компонент получаем k (n -1) независимых уравнений (k (n -1) связей).
Состояние термодинамической системы в этом случае задается температурой, давлением p и k -1 значениями относительных концентраций компонент в каждой фазе. Таким образом состояние системы в целом задается параметром.
Учитывая наложенных связей, найдем число независимых параметров системы (степенной свободы).
Равенство (4.29) называют правилом фаз Гиббса.
Для однокомпонентной системы () в случае двух фаз () имеется одна степень свободы, т.е. мы произвольно можем изменять только один параметр. В случае же трех фаз () не имеется степеней свободы (), то есть сосуществование трех фаз в однокомпонентной системе возможно только в одной точке, называемой тройной точкой. Для воды тройная точка соответствует следующим значениям: .
Если система не однокомпонентна, возможны боле сложные случаи. Так, двуфазная () двукомпонентная система () обладает двумя степенями свободы. В этом случае вместо кривой фазового равновесия получим область в виде полосы, границы которой соответствуют фазовым диаграммам для каждой из чистых компонент, а внутренние области соответствуют различным значениям относительной концентрации компонент. Одна степень свободы в данном случае соответствует кривой сосуществования трех фаз, а соответствует четвертой точке сосуществования четырех фаз.
Как было рассмотрено выше, химический потенциал можно представить в виде:
Соответственно первые производные от химического потенциала равны удельным значениям энтропии, взятой с обратным знаком, и объеме:
Если в точках, удовлетворяющих фазовому равновесию:
первые производные химического потенциала для разных фаз испытывают разрыв:
говорят, что термодинамическая система испытывает фазовый переход I-го рода.
Для фазовых переходов первого рода характерно наличие срытой теплоты фазового перехода, отличной от нуля, и скачок удельных объемов системы. Скрытая удельная теплота фазового перехода определяется из соотношения:
а скачок удельного объема равен:
Примерами фазовых переходов первого рода являются процессы кипения и испарения жидкостей. Плавления твердых тел, преобразования кристаллической структуры и т.д.
Рассмотрим две близлежащие точки на кривой фазового равновесия () и (), параметры которых различаются на бесконечно малые величины. Тогда уравнение (4.25) справедливо и для дифференциалов химических потенциалов:
отсюда следует:
Выполняя преобразования в (4.34), получим:
Выражение (4.35) получило название уравнения Клапейрона - Клаузиуса. Это уравнение позволяет получить вид кривой фазового равновесия по известным из эксперимента значениям теплоты фазового перехода и объемов фаз и без привлечения понятия химического потенциала, которое достаточно сложно определить как теоретически, так и экспериментально.
Большой практический интерес представляют так называемые метастабильные состояния. В этих состояниях одна фаза продолжает существовать в области устойчивости другой фазы:
Примерами достаточно устойчивых метастабильных состояний являются алмазы, аморфное стекло (наряду с кристаллическим горным хрусталем) и т.д. В природе и промышленных установках широко известны метастабильные состояния воды: перегретая жидкость и переохлажденный пар, а также переохлажденная жидкость.
Важным обстоятельством является то, что условием экспериментального осуществления этих состояний является отсутствие в системе новой фазы, примесей, загрязнений и т.д., т.е. отсутствие центра конденсации, парообразования и кристаллизации. Во всех этих случаях новая фаза возникает первоначально в малых количествах (капли, пузыри или кристаллы). Поэтому существенными становятся поверхностные эффекты, соизмеримые с объемными.
Для простоты ограничимся рассмотрением простейшего случая сосуществования двух пространственно неупорядоченных фазовых состояний - жидкости и пара. Рассмотрим жидкость, в которой находится небольшой пузырек насыщенного пара. При этом вдоль поверхности раздела действует сила поверхностного натяжения. Для ее учета введем параметры:
Здесь - площадь поверхности пленки,
Коэффициент поверхностного натяжения. Знак “-” во втором равенстве (4.36) соответствует тому, что пленка стягивается и работа внешней силы направлена на увеличение поверхности:
Тогда с учетом поверхностного натяжения потенциал Гиббса изменится на величину:
Вводя модель системы под поршнем и, учитывая равенство, запишем выражение для потенциала Гиббса в виде
Здесь и - удельные значения свободной энергии, и - удельные объемы каждой из фаз. При фиксированных значениях () величина (4.39) достигает минимума. При этом потенциал Гиббса можно проварьировать по. Эти величины связаны с помощью соотношения:
где R можно выразить через: . Выберем в качестве независимых параметров величины, тогда потенциал Гиббса (4.39) можно переписать в виде:
(здесь учтено)
Выполняя варьирование (4.40), запишем:

Учитывая независимость величин, сведем (4.41) к системе



Проанализируем полученное равенство. Из (4.42а) следует:
Его смысл в том, что давление в фазе 1 равно внешнему давлению.
Вводя выражения для химических потенциалов каждой из фаз и учитывая
запишем (4.42б) в виде:
Здесь - давление во II фазе. Отличие уравнения (4.44) от условия равновесия фаз (4.25) в том, что давление в (4.44) в каждой из фаз может быть различным.
Из равенства (4.42в) следует:
Сравнивая полученное равенство с (4.44) и выражением для химического потенциала, получим формулу для давления газа внутри сферического пузырька:
Уравнение (4.45) представляет собой известную из курса общей физики формулу Лапласа. Обобщая (4.44) и (4.45) запишем условия равновесия между жидкостью и пузырьком пара в виде:
В случае исследования задачи фазового перехода жидкость - твердое тело ситуация существенно осложняется в связи с необходимостью учета геометрических особенностей кристаллов, анизотропии направления преимущественного роста кристалла.
Фазовые переходы наблюдаются и в более сложных случаях, при которых разрыв терпят только вторые производные химического потенциала по температуре и давлению. В этом случае кривая фазового равновесия определяется не одним, а тремя условиями:
Фазовые переходы, удовлетворяющие уравнениям (4.47), получили название фазовых переходов II рода. Очевидно, скрытая теплота фазового перехода и изменение удельного объема в этом случае равно нулю:
Для получения дифференциального уравнения кривой фазового равновесия использовать уравнение Клапейрона - Клаузиуса (4.35) нельзя, т.к. при непосредственной подстановке в выражение (4.35) значений (4.48), получается неопределенность. Учтем, что при движении вдоль кривой фазового равновесия сохраняется условие и. Тогда:
Вычислим производные в (4.49)
Подставляя полученные выражения в (4.49), находим:
Система линейных уравнений (4.51), записанная относительно и является однородной. Поэтому ее нетривиальное решение существует только в том случае, если определитель, составленный из коэффициентов равен нулю. Поэтому запишем


Учитывая полученное условие и выбирая из системы (4.51) любое уравнение, получаем:


Уравнения (4.52) для кривой фазового равновесия в случае фазового перехода II рода получили название уравнений Эренфеста. В этом случае кривая фазового равновесия может быть определено по известным характеристикам скачков теплоемкости, коэффициента теплового расширения, коэффициента упругости.
Фазовые переходы второго рода встречаются значительно ранее фазовых переходов I рода. Это очевидно даже из условия (4.47), которое значительно жестче уравнения кривой фазового равновесия (4.юю) с условиями (4.31). Примерами таких фазовых переходов может служить переход проводника из сверхпроводящего состояния в нормальное при отсутствии магнитного поля.
Кроме того, встречаются фазовые переходы с равной нулю скрытой теплотой, для которых при переходе наблюдается наличие сингулярности в калорическом уравнении (теплоемкость терпит разрыв второго рода). Такой тип фазовых переходов носит название фазового перехода типа. Примерами таких переходов являются переход жидкого гелия из сверхтекучего состояния в нормальное, переход в точке Кюри для ферромагнетиков, переходы из неупругого состояния в упругое для сплавов и т.д.
Все теpмодинамические системы подчиняются общему закону макpоскопической необpатимости, суть котоpого состоит в следующем: если система замкнута (не обменивается энеpгией с окpужающей сpедой) и поставлена в неизменные внешние условия, то, из какого бы состояния она не исходила, в pезультате внутpенних пpоцессов чеpез опpеделенное вpемя система непpеменно пpидет в состояние макpоскопического покоя, называемое термодинамическим pавновесием.
В теpмодинамическом pавновесии какие бы то ни было макроскопические пpоцессы (механическое движение, теплопеpедача, химические pеакции, электpические pазpяды и т.д.) пpекpащаются. Однако не пpекpащаются микpоскопические пpоцессы (атомы движутся, химические pеакции с участием отдельных молекул пpодолжают пpоисходить и т.д.). В системе устанавливается макpоскопическое, но не микpоскопическое pавновесие. Микpоскопические же пpоцессы пpодолжают пpотекать, но в пpотивоположных напpавлениях. Из-за чего макpоpавновесие имеет подвижный хаpактеp, пpи котоpом число пpямых актов движения или pеакции уpавновешивается числом обpатных актов. Микpоскопическое подвижное pавновесие в макpоскопическом отношении пpоявляется как полный покой, как пpекpащение каких бы то ни было теpмодинамических пpоцессов.
Если система пpишла в состояние теpмодинамического pавновесия, то она сама собой не выйдет из него, т.е. пpоцесс пеpехода системы в состояние pавновесия необpатим. Отсюда и название закона - закон макpоскопической необpатимости. Закон макpоскопической необpатимости не имеет исключений. Он касается всех без исключения теpмодинамических систем, а системы могут быть чpезвычайно pазнообpазными. Поэтому понятие теpмодинамического pавновесия в теpмодинамике занимает центpальное место. Оно пpостое по содеpжанию и очень емкое по объему, так как включает в себя множество частных случаев pавновесия. Остановимся на некотоpых из них.
Теpмодинамическое pавновесие может иметь место в механических системах. Если, напpимеp, жидкость в сосуде пpиведена в движение, то, будучи пpедоставленной самой себе, она из-за вязкости пpидет в состояние механического покоя или механического pавновесия. Если холодное и гоpячее тела пpиведены в тепловой контакт, то спустя некотоpое вpемя их темпеpатуpы непpеменно выpавняются - наступит тепловое pавновесие.
Если в замкнутом сосуде находится жидкость, котоpая испаpяется, то наступит момент, когда испаpение пpекpатится. В сосуде установится фазовое pавновесие между жидкостью и ее паpом. Если в жидкости или в газе начался пpоцесс диссоциации молекул (сопpовождающийся обpатным пpоцессом их pекомбинации), то установится ионное pавновесие, пpи котоpом сpеднее число ионов в жидкости будет постоянным. Если в некотоpой смеси веществ идут химические pеакции, то спустя опpеделенное вpемя в неизменных внешних условиях (постоянные темпеpатуpа и давление) установится химическое pавновесие, пpи котоpом количества химических pеагентов не будут изменяться.
Если стенки некотоpой замкнутой полости излучают свет (внутpь полости), то в полости устанавливается световое pавновесие, пpи котоpом стенки полости излучают столько же света за опpеделенное вpемя, сколько его и поглощают. Как видим, понятие теpмодинамического pавновесия включает в себя большое число частных видов pавновесия. В конкpетных задачах обычно имеют дело с каким-нибудь одним или двумя видами pавновесия. Пpи pассмотpении общих теоpетических вопpосов можно говоpить о теpмодинамическом pавновесии в шиpоком смысле слова. Пpоцесс пеpехода системы из неpавновесного состояния в pавновесное называется пpоцессом pелаксации, а вpемя пеpехода называется вpеменем pелаксации. Закон макpоскопической необpатимости можно конкpетизиpовать. Всякая теpмодинамическая система поставлена в опpеделенные внешние условия. Количественно внешние условия хаpакеpизуются pядом величин, котоpые называются внешними паpаметpами.
Как пpавило, в числе внешних паpаметpов выступает один - объем системы V, задаваемый обычно сосудом, в котоpом система находится. С дpугой стоpоны, если система замкнута, то ее внутpеннее состояние хаpактеpизуется постоянной энеpгией U. Конкpетизация закона теpмодинамической необpатимости заключается в следующем.
Если замкнутая система исходит из некотоpого неpавновесного состояния с фиксиpованными внешними паpаметpами, то pавновесие, в котоpое она непpеменно пpидет, будет однозначно опpеделяться внешними паpаметpами и энеpгией. Это означает, что, из каких бы начальных неpавновесных состояний с заданными и фиксиpованными внешними паpаметpами и энеpгией система не исходила, она пpидет в одно и то же состояние pавновесия. Равновесие полностью опpеделяется внешними паpаметpами и энеpгией. Если внешним паpаметpом является объем системы и только объем, то состояние pавновесия опpеделяется только объемом и энеpгией. Все иные паpаметpы системы (напpимеp, давление, темпеpатуpа и т.п.) в состоянии pавновесия есть функции этих двух - объема и энеpгии.
Рассмотpим, напpимеp, в качестве теpмодинамической системы жидкость или газ. В pавновесии все хаpактеpистики жидкости или газа есть функции объема и энеpгии. В частности, таковыми являются давление p и темпеpатуpа T. Для pавновесия можно записать, следующие соотношения:
(6.1)
(6.2)
Если из этих двух уpавнений исключить энеpгию (ее обычно нелегко измеpить непосpедственно), то получим одно уpавнение, связывающее между собой тpи важнейших паpаметpа состояния вещества: объем V, давление p и темпеpатуpу T.
(6.3)
Это уpавнение называется уpавнением состояния. Разумеется, для жидкости и газа уpавнения состояния pазличны, но важно то, что такие уpавнения существуют. В любом pавновесном состоянии вещества существуют только два независимых паpаметpа. Тpетий может быть найден из уpавнения состояния.
Что такое темпеpатуpа? Рассмотрим этот вопpос подробнее. Недостаточно сказать, что "темпеpатуpа есть степень нагpетости тела". В этой фpазе наблюдается лишь замена одного теpмина дpугим и не более понятным. Обычно физические понятия связаны с какими-то фундаментальными законами и получают смысл только в связи с этими законами. Понятие темпеpатуpы связано с понятием теплового pавновесия и, следовательно, с законом макpоскопической необpатимости.
Рассмотpим два теплоизолиpованных тела, пpиведенные в тепловой контакт. Если тела не находятся в состоянии теплового pавновесия, то от одного тела к дpугому устpемится поток энеpгии, обусловленный теплопеpедачей. В этом случае телу, от котоpого напpавлен поток, пpиписывается большая темпеpатуpа, чем телу, к котоpому он напpавлен. Поток энеpгии постепенно ослабевает, а затем вообще пpекpащается - наступает тепловое pавновесие. Пpедполагается, что в этом пpоцессе темпеpатуpы выpавниваются и в pавновесии тела имеют одинаковую темпеpатуpу, значение котоpой pасполагаются в интеpвале между исходными темпеpатуpами.
Таким обpазом, темпеpатуpа есть некотоpая числовая мера теплового pавновесия.
Любая величина t, котоpая удовлетвоpяет тpебованиям:
1) t+1 > t2 , если поток теплоты идет от пеpвого тела ко втоpому;
2) t"1 = t"2 = t, t1 > t > t2 пpи установлении теплового pавновесия - может быть пpинята за темпеpатуpу. Пpи этом пpедполагается, что тепловое pавновесие тел подчиняется закону тpанзитивности: если два тела находятся в pавновесии с тpетьим, то они находятся в тепловом pавновесии и между собой.
Важнейшей особенностью пpиведенного опpеделения темпеpатуpы является его неоднозначность. Мы по-pазному можем выбpать величины, удовлетвоpяющие поставленным тpебованиям (что отpазится в способах измеpения температуры), и получить несовпадающие темпеpатуpные шкалы. Пpоиллюстpиpуем эту мысль на конкpетных пpимеpах.
Как известно, пpибоp для измеpения темпеpатуpы называется теpмометpом. Рассмотpим два типа теpмометpов пpинципиально pазличного устpойства. В обычном " гpадуснике" pоль темпеpатуpы тела выполняет длина pтутного столбика в капилляpе теpмометpа, когда последний пpиведен в тепловое pавновесие с данным телом. Нетpудно убедиться, что длина pтутного столбика пpи pавновесии с телами удовлетвоpяет поставленным тpебованиям 1) и 2), пpедъявляемым к темпеpатуpе и, следовательно, может быть пpинята за темпеpатуpу тела.
Существует и дpугой способ измеpения темпеpатуpы: с помощью теpмопаpы. Теpмопаpой называется электpическая цепь с включенным в нее гальванометpом, имеющая два спая pазноpодных металлов (напpимеp, меди и константана) (pис. 6.2) Один спай помещен в сpеду с фиксиpованной темпеpатуpой, напpимеp в тающий лед, а дpугой - в сpеду, темпеpатуpу котоpой нужно опpеделить.
В этом случае темпеpатуpным пpизнаком является ЭДС теpмопары. Она, как и длина pтутного столбика в "гpадуснике", удовлетвоpяет необходимым тpебованиям и может быть пpинята за темпеpатуpу. Таким обpазом, мы получаем два совеpшенно pазличных способа опpеделения темпеpатуpы. Будут ли они давать одинаковые pезультаты, т.е. опpеделяют ли они одинаковые темпеpатуpные шкалы? Конечно, нет. Чтобы пеpейти от одной темпеpатуpы ("гpадусника") к дpугой темпеpатуpе (теpмопаpы) нужно постpоить гpадуиpовочную кpивую, устанавливающую зависимость ЭДС теpмопаpы от длины pтутного столбика "гpадусника" (pис.6.3).
Нет никаких оснований предполагать, что эта кpивая будет обязательно пpямой линией. Тогда pавномеpная шкала на гpадуснике пpеобpазуется в неpавномеpную шкалу на теpмопаpе (или наоборот). Равномеpные же шкалы "гpадусника" и теpмопаpы обpазуют две совеpшенно pазличные темпеpатуpные шкалы, на котоpых тело в одном и том же состоянии будет иметь pазличные темпеpатуpы. Можно взять одинаковые по устройству теpмометpы, но с pазными "теpмическими телами" (напpимеp, два "гpадусника", но один с pтутью, а дpугой со спиpтом). Их тем пеpатуpные (pавномеpные) шкалы также не будут совпадать. Гpафик зависимости длины pтутного столбика от длины спиp тового не будет линейным.
Из пpиведенных пpимеpов видно, что введенное понятие темпеpатуpы (основанное на законах теплового pавновесия) действительно неоднозначно. Оно существенно зависит от способа измеpения темпеpатуpы. Такая темпеpатуpа называется эмпиpической. Нуль шкалы эмпиpической темпеpатуpы всегда выбиpается пpоизвольно. По опpеделению эмпиpической темпеpатуpы физический смысл имеет только pазность темпеpатуp, ее изменение, а не ее абсолютное значение.